
Oxycodone, hydrocodone, morphine, and other opioids are the most effective analgesic medications currently available, but they can be addictive and lethal when misused. The United States is currently experiencing an epidemic of such misuse; treatment admissions for abuse of prescription opioids quadrupled from 1998 to 2008, and opioid overdose was second only to road accidents as a cause of unintentional deaths in 2009. As part of NIDA’s comprehensive efforts to curb this epidemic, investigators are searching for safer analgesic alternatives. A NIDA-supported group at the University of California, Irvine, have developed a new compound that sharply reduced rats’ responses in experimental models of pain, including some for which opioids are sometimes the only effective remedy.
“The compound has a completely new mechanism,” says lead investigator Dr. Daniele Piomelli. “We do not yet know whether it will be safe and effective in people. However, it is effective in animals and definitely has features that are good for a pain medication. It does not enter the brain, so it can’t produce the central nervous system (CNS) effects that cause problems with opioids.” Instead, the compound enhances and prolongs the activity of a neurotransmitter that dampens pain signals at their source in peripheral tissues.
Tackling Pain at Its Source
Pain is a protective response, signaling the need to react to conditions that stress or damage tissues—for example, to withdraw from an injurious contact or protect an injured body part from further insults. Pain that is too great or lasts too long is counterproductive, however, so the body generates counter-responses aimed at tempering and ultimately eliminating pain episodes. When these antinociceptive (pain-reducing) responses fall short, pain can persist and become chronic.
Morphine, oxycodone, and other opioids tap into the best known of the body’s antinociceptive responses. They activate the same receptors that the body’s natural opioid peptides stimulate to dampen signaling in brain pathways that contribute to pain perception. Opioids can relieve pain that does not respond to other medications, but they also can cause CNS side effects including drowsiness, respiratory depression, euphoria, physical tolerance, and addiction.
The compound developed by Dr. Piomelli and colleagues modulates a different antinociceptive response. In 1998, Dr. Piomelli and colleagues suggested that anandamide, a neurotransmitter in the cannabinoid system, blocks pain perception by binding to receptors on sensory nerves outside of the CNS. Studies confirmed the hypothesis. In one, the researchers eliminated cannabinoid receptors from the peripheral tissues of mice through a genetic manipulation. The mice exhibited heightened sensitivity to pain originating in their extremities, even though they still had functioning cannabinoid receptors in the CNS.
Dr. Piomelli says that the finding showed that, “The cannabinoid receptors outside the brain provide a filter for pain.” This being so, compounds that enhance stimulation of peripheral cannabinoid receptors might confer analgesia without the downsides of opioids.
The UC group’s compound, called URB937, increases anandamide activity at peripheral cannabinoid receptors in an indirect manner. It inhibits the activity of an enzyme called fatty acid amine hydrolase (FAAH), which breaks down anandamide. As a result, anandamide molecules that are released as part of the body’s antinociceptive response remain intact and activate cannabinoid receptors for a longer period.
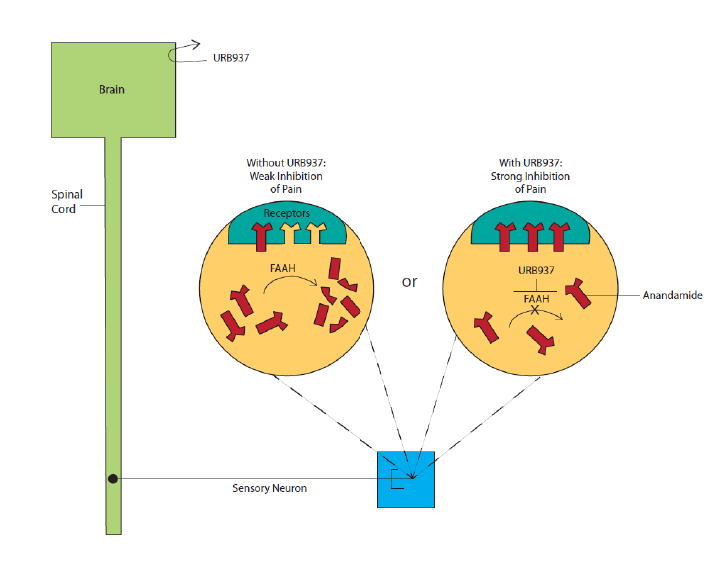 Mechanism of Action of a New Pain Reliever URB937 blocks the enzyme fatty acid amide hydrolase (FAAH) from degrading anandamide in peripheral tissues. Anandamide accordingly accumulates, and, when it binds to receptors, inhibits the transmission of pain signals to the spinal cord and brain.
Mechanism of Action of a New Pain Reliever URB937 blocks the enzyme fatty acid amide hydrolase (FAAH) from degrading anandamide in peripheral tissues. Anandamide accordingly accumulates, and, when it binds to receptors, inhibits the transmission of pain signals to the spinal cord and brain.Because URB937 only blocks FAAH in peripheral tissues, it reinforces the neurotransmitter’s peripheral pain-relieving efficacy without disrupting its central effects. In fact, the compound appears to concentrate or confine its impact to the immediate vicinity of tissue damage: In a previous study, URB937 reduced rats’ sensitivity to heat and pressure applied to an injured forepaw, but not to the forepaw on the other side of their body.
Strategy, Diligence, Luck
The UC researchers’ strategy for developing URB937 was to modify FAAH inhibitors they had previously discovered, which entered the brain. The modification made the inhibitors more hydrophilic, or interactive with water. Endothelial cells that line capillaries in the brain restrict the passage of hydrophilic substances from the blood into the cerebrospinal fluid.
The group synthesized and tested several hydrophilic FAAH inhibitors before hitting upon URB937. The early versions penetrated the endothelial membrane and inhibited brain FAAH activity despite their hydrophilicity. Despite being only slightly different from the others, URB937 was completely excluded from the CNS (see figure). “This is where we got lucky,” says Dr. Piomelli. “A simple modification did not just reduce the amount of compound that got through, it completely eliminated entry into the brain.
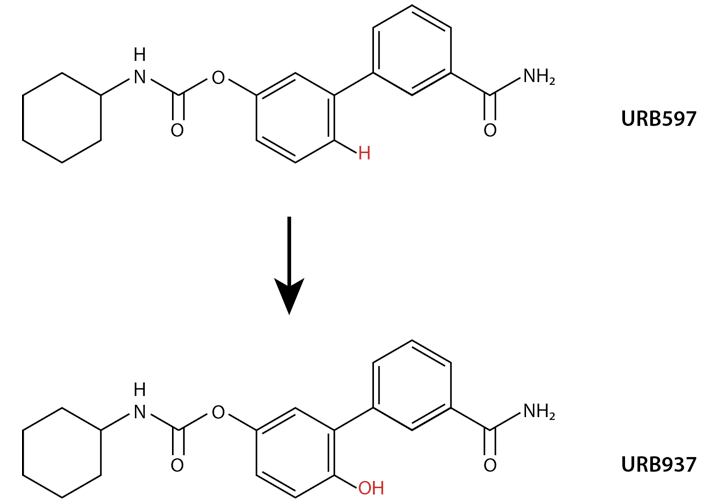 Inhibitor That Acts Only in the Peripheral Nervous System The FAAH inhibitor URB937 was designed by making a simple chemical modification to a previously designed FAHH inhibitor (URB597), which acts both in the central nervous system and the periphery. The modification puts a hydroxyl group (OH) in the place of a hydrogen (H).
Inhibitor That Acts Only in the Peripheral Nervous System The FAAH inhibitor URB937 was designed by making a simple chemical modification to a previously designed FAHH inhibitor (URB597), which acts both in the central nervous system and the periphery. The modification puts a hydroxyl group (OH) in the place of a hydrogen (H).At first the researchers were puzzled that the small chemical change that converted a previous FAAH inhibitor to URB937 could have such a decisive effect. They then discovered that besides making the FAAH molecule more hydrophilic, their molecular tweak had inadvertently exposed URB937 to binding by a transporter protein on the surface of the endothelial membrane. This transporter captures substances that intersect the membrane and carries some into the cell and others—including URB937—out.
Dr. Piomelli and colleagues identified the specific transporter that keeps URB937 out of the CNS. This well-studied protein, called ABCG2, also known as the breast cancer resistance protein, plays a significant role in drug distribution. Now that scientists have identified this mechanism, they will be better able to design additional FAAH inhibitors that only affect the peripheral nervous system.
Pains Tamed
In the latest tests of URB937, Dr. Piomelli and colleagues gave the drug orally to rats and mice in protocols designed to simulate human acute inflammation, peripheral neurogenic pain, and arthritic pain. In the first protocol, mice received a solution of URB937 or an inert substance, followed by an injection of the chemical irritant carrageenan into a hind paw. All the mice developed swelling of the injected paw, which was most marked in the animals treated with the inert substance and least marked in those receiving the highest dose of URB937 (3 mg/kg of body weight).
Mice also exhibited behavioral evidence that URB937 reduced two other accompaniments to inflammation: increased sensitivity to pain from heat and mechanical pressure. Animals treated with URB937 withstood more radiant heat from a focused beam and endured steady pressure from a pointed glass cylinder for a longer time before removing the injected paw (see Compound Suppresses Acute Inflammatory Pain). Again, higher doses of URB937 were associated with increased tolerance. A 3 mg/kg dose curbed swelling and both types of pain for 48 hours.
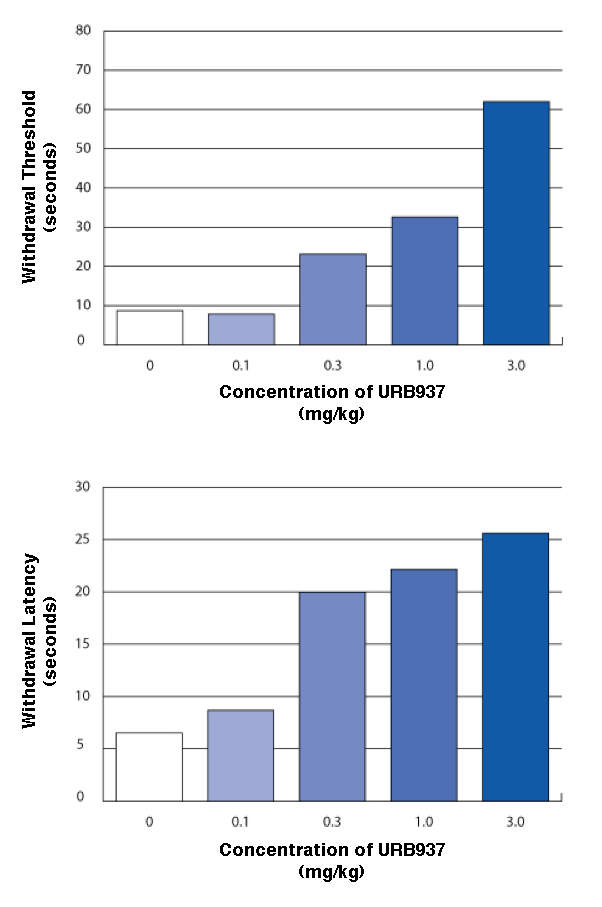 Compound Suppresses Acute Inflammatory Pain In a protocol designed to simulate acute inflammatory pain from arthritis, mice received a hind-paw injection of the chemical irritant carrageenan, which resulted in swelling. Animals that received oral URB937, as compared to those that did not receive the compound, withstood more radiant heat from a focused beam before withdrawing the swollen paw (Withdrawal Threshold) and withstood longer periods of steady pressure on the paw from a pointed glass cylinder (Withdrawal Latency). Higher doses of URB937 were associated with increased pain tolerance.
Compound Suppresses Acute Inflammatory Pain In a protocol designed to simulate acute inflammatory pain from arthritis, mice received a hind-paw injection of the chemical irritant carrageenan, which resulted in swelling. Animals that received oral URB937, as compared to those that did not receive the compound, withstood more radiant heat from a focused beam before withdrawing the swollen paw (Withdrawal Threshold) and withstood longer periods of steady pressure on the paw from a pointed glass cylinder (Withdrawal Latency). Higher doses of URB937 were associated with increased pain tolerance.
- Text description
-
Top graph: Bar graph compares how long mice with acute inflammation took to withdraw their paws from a hot surface when untreated and when treated with four doses of URB937. The more URB937 animals received, the longer they left their paws on the surface, from a minimum of 10 seconds (untreated) to a maximum of 60 seconds (3.0 mg/kg).
Bottom graph: Bar graph compares how long mice with acute inflammation took to withdraw their paws from a radiant heat source when untreated and when treated with four doses of URB937. The more URB937 animals received, the longer they left their paws on the surface, from a minimum of 5 seconds (untreated) to a maximum of 25 seconds (3.0 mg/kg).
Severe chronic pain requiring long-term treatment with opioids commonly occurs when nerves are injured. Using a conventional protocol for producing such pain, the researchers surgically (and reversibly) exposed animals’ sciatic nerve and tied a fiber around it. After 3 days, when the pain had essentially become chronic, the mice were given URB937 or an inert substance. Two hours later, the mice that received 3 mg/kg of URB937 showed no more sensitivity to fixed-intensity radiant heat or pressure from a glass cylinder than control animals whose nerves were not tied (see Chronic Neurogenic Pain Responds to URB937). URB937 also attenuated another consequence of nerve damage, pain felt in response to touching that ordinarily does not hurt.
 Chronic Neurogenic Pain Responds to URB937 Seven days after having their sciatic nerve surgically exposed and tied, mice received either an inert substance or URB937. Two hours later, those that received 3 mg/kg of the FAAH inhibitor showed no more sensitivity to radiant heat (Withdrawal Threshold) or pressure from a glass cylinder (Withdrawal Latency) than control animals whose nerves were not tied (Sham).
Chronic Neurogenic Pain Responds to URB937 Seven days after having their sciatic nerve surgically exposed and tied, mice received either an inert substance or URB937. Two hours later, those that received 3 mg/kg of the FAAH inhibitor showed no more sensitivity to radiant heat (Withdrawal Threshold) or pressure from a glass cylinder (Withdrawal Latency) than control animals whose nerves were not tied (Sham).
- Text description
-
Top diagram: Bar graph compares how long mice with simulated chronic neurogenic pain took to withdraw their paws from a hot surface when untreated and when treated with four doses of URB937. The more URB937 animals received, the longer they left their paws on the surface, from a minimum of 10 seconds (untreated) to a maximum of 45 seconds (3.0 mg/kg).
Bottom diagram: Bar graph compares how long mice with simulated chronic neurogenic pain took to withdraw their paws from a radiant heat source when untreated and when treated with four doses of URB937. The more URB937 animals received, the longer they left their paws on the surface, from a minimum of 15 seconds (untreated) to a maximum of 48 seconds (3.0 mg/kg).
The FAAH inhibitor also alleviated animals’ heightened pain responses to heat and pressure in a protocol that produces chronic inflammation resembling arthritis. The researchers gave mice URB937, an inert substance, or dexamethasone, a medication commonly prescribed for arthritis, 3, 7, or 14 days after administering the inflammation-causing agent called complete Freund’s adjuvant. On each day, the highest dose tested (30 mg/kg) of URB937 reduced pain behavior as much as 100 mg/kg of dexamethasone.
Dr. Piomelli and colleagues also found evidence that URB937 may reduce dosing requirements for indomethacin, a widely prescribed anti-inflammatory medication that has an undesirable side effect of promoting gastric ulcers. In the acute pain protocol, doses of the two drugs that were ineffective when given individually markedly reduced tissue swelling and pain responses to heat and pressure when given together. Moreover, food-deprived animals that received both URB937 and indomethacin developed less gastric irritation than those that received indomethacin alone.
Dr. Piomelli is currently seeking a private partner interested in further developing URB937 and related chemicals. “We are working really hard to move this compound toward the clinic,” he says.
The work with URB937 builds on earlier NIDA-funded studies to develop analgesics that target the peripheral nerves. “Eventually we want to eliminate morphine and other opioids as pain medications,” says Dr. Rao Rapaka, chief of NIDA’s Chemistry and Physiological Systems Research Branch. “In addition to Dr. Piomelli’s promising results with FAAH inhibition, his method for designing molecules that do not get into the central nervous system can have broad application in the design of other medications.”
Sources
Clapper, J.R., et al. Anandamide suppresses pain initiation through a peripheral endocannabinoid mechanism. Nature Neuroscience 13(10): 1265–1270, 2010. Full Text
Lovinger, D.M. Endocannabinoids rein in pain outside the brain. Nature Neuroscience 13(10): 1155–1156, 2010. Full Text
Moreno-Sanz, G., et al. The ABC membrane transporter ABCG2 prevents access of FAAH inhibitor URB937 to the central nervous system. Pharmacological Research 64(4):359–363, 2011. Abstract
Sasso, O., et al. Peripheral FAAH inhibition causes profound antinociception and protects against indomethacin-induced gastric lesions. Pharmacological Research 65(5): 553–563, 2012. Abstract