
- Two novel compounds powerfully suppressed animals’ pain responses, while producing little or none of the respiratory depression and liability for misuse and abuse associated with morphine and other typical opioids.
- One of the compounds was extensively tested in a monkey model, laying key groundwork for moving to human trials.
- The other compound was identified using computational modeling based on x-ray crystallography of a receptor’s structure, and exemplifies that technique’s potential for identifying novel compounds that can meet highly specific therapeutic needs.
Two new studies bode well for the prospect of developing highly effective pain treatments without the risks for misuse and potentially fatal overdose associated with typical opioids. Although both the compounds evaluated in both studies work through the µ opioid receptor (MOR), only one of them has a classical opioid ligand structure, while the other is chemically novel. Both preserve the analgesia conferred by stimulating MOR while skirting some of the associated dose-limiting side effects.
Compound’s Dual Action Suppresses Pain in Monkeys
Dr. Huiping Ding and colleagues from the Department of Physiology and Pharmacology at the Wake Forest University School of Medicine and the Department of Pharmacy and Pharmacology at the University of Bath, United Kingdom, previously showed that a compound, BU08028, reduces rats’ pain responses as effectively as morphine. They have now tested BU08028 in rhesus macaque monkeys, which is the next step in determining whether it is safe and sufficiently promising to warrant giving it to people in a clinical trial.
BU08028 stimulates the MOR on neurons, and it also stimulates a second opioid receptor, called the nociceptin FQ receptor (NOR). Both types of stimulation suppress pain, but of the two, only MOR stimulation causes respiratory side effects and reinforcement that motivates abuse and addiction. In theory, BU08028’s NOR stimulation will control a portion of the patient’s pain; as a result, less MOR stimulation will be needed to control the remaining pain, and the side effects related to MOR stimulation will be reduced accordingly. NOR stimulation also may directly counteract opioids’ reinforcing effect.
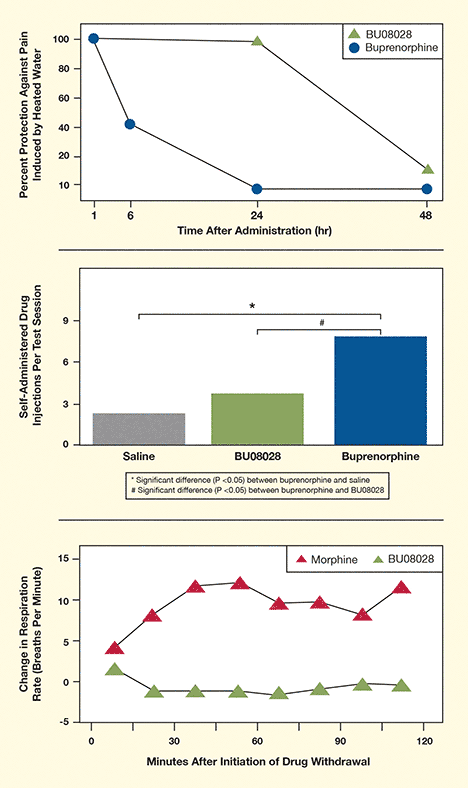 Figure 1. B0U8028 Reduces Pain but Causes Neither Reinforcement nor Withdrawal Top panel: BU08028 provided monkeys with relief from a painful heat stimulus for three times longer than the opioid buprenorphine. Middle panel: Monkeys self-administered fully pain-suppressing and higher doses of BU08028 no more often than they did saline solution, indicating that BU08028 had no reinforcing effect. Bottom panel: Monkeys exhibited increased respiration rates, a sign of withdrawal, when morphine was suddenly withdrawn, but not when BU08028 was withdrawn.
Figure 1. B0U8028 Reduces Pain but Causes Neither Reinforcement nor Withdrawal Top panel: BU08028 provided monkeys with relief from a painful heat stimulus for three times longer than the opioid buprenorphine. Middle panel: Monkeys self-administered fully pain-suppressing and higher doses of BU08028 no more often than they did saline solution, indicating that BU08028 had no reinforcing effect. Bottom panel: Monkeys exhibited increased respiration rates, a sign of withdrawal, when morphine was suddenly withdrawn, but not when BU08028 was withdrawn.
- Text Description of Figure 1
-
The figure has three panels of graphs comparing the effectiveness, reinforcing effects, and withdrawal effects of a new opioid compound, BU08028, with that of buprenorphine and morphine. The top panel shows two graphs representing the effectiveness of BU08028and buprenorphine in protecting against pain in monkeys. The vertical y-axis indicates the percent protection against pain induced by heated water. The horizontal x-axis indicates the time after drug administration in hours, with 1, 6, 24, and 48 hours marked. Values for BU08028 are indicated by green triangles; values for buprenorphine are indicated by blue circles. Both buprenorphine and BU08028show 100 percent protection against pain at 1 hour after administration. For buprenorphine, the protection against pain decreases to slightly more than 40 percent after 6 hours and then further to less than 10 percent after 24 and 48 hours. For BU08028, protection against pain remains at nearly 100 percent after 24 hours, before declining to around 15 percent after 48 hours, indicating much greater and prolonged pain protection compared with buprenorphine.
The middle panel is a bar chart comparing reinforcing effects of BU08028 and buprenorphine as indicated by drug self-administration in monkeys. The vertical y-axis indicates the number of self-administered drug injections the animals performed per test session, with a scale of 3, 6, or 9 injections. As a negative control, the gray left bar shows self-administered injections after treatment with a saline solution, which was approximately 2 injections per session. The green middle bar shows that the number of self-administered injections after treatment with BU08028 was slightly higher at about 3.5 injections per session. The blue right bar shows that the number of self-administered injections after treatment with buprenorphine was about 8 injections per session. A horizontal line above the bars from saline to buprenorphine with an asterisk indicates that the difference between buprenorphine and saline was statistically significant with a P value of less than 0.06. A horizontal line between the bars from BU08028 to buprenorphine with a hash sign indicates that the difference between buprenorphine and BU08028 was statistically significant with a P value of less than 0.06, indicating that BU08028 has a significantly lower reinforcing effects than buprenorphine.
The bottom panel shows two line graphs comparing the withdrawal effects of BU08028 and morphine. The vertical y-axis shows the change in respiration rate as breaths per minute after sudden drug withdrawal. The scale ranges from -5 to +15. The horizontal x axis shows the time in minutes after drug withdrawal, with 0, 30, 60, 90, and 120 minutes indicated. Values for BU08028 are indicated by green triangles; values for morphine are indicated by red triangles. The line graph for BU08028 shows a slight increase in respiration of about 1.5 breaths per minute during the first 15 minutes after drug withdrawal; after that, the respiration rate returns to baseline or even slightly below baseline and remains at that level throughout the test period. The line graph for morphine shows an increase in respiration of about 4 breaths per minute during the first 15 minutes after withdrawal initiation, an increase of about 8 breaths per minute during the second 15 minutes, and increases of about 9 to 11 breaths per minute during the remainder of the test period. This indicates that morphine withdrawal induced withdrawal effects, whereas BU08028 withdrawal induced no such effects.
In the tests, the monkeys’ pain responses to heated water (120 °F) and to simulated pain hypersensitivity completely resolved when they were treated systemically with BU08028. Dose for dose, BU08028 provided stronger pain relief than the partial opioid agonist buprenorphine and lasted longer—more than 24 hours, as opposed to 1–6 hours (see Figure 1). Given the opportunity to self-administer BU08028 at fully pain-suppressing doses or at doses that were 10 or more times higher, the monkeys exhibited no greater motivation to do so than to self-administer saline solution. At the fully pain-suppressing dose, comprehensive measurements found no diminution of the animals’ respiratory or cardiovascular function. Administration of an opioid antagonist produced signs of withdrawal after animals were exposed for 3 days to morphine, but not to BU08028.
Dr. Mei-Chuan Ko, lead author of the study report, says the study’s results advance hopes that BU08028 might become an effective, low-risk alternative to morphine and other current prescription opioid analgesics. “Rhesus monkeys could serve as a surrogate species for humans in the development of opioid-related [medications], as several behavioral and physiological assays in nonhuman primates have been demonstrated to have predictive validity for the clinical use of opioids,” he says.
Although the results of the self-administration studies suggest that BU08028 is less reinforcing than cocaine and other drugs, more data will be required to ascertain that this compound lacks liability for misuse and abuse. The researchers also stress the need for studies using higher BU08028 doses and longer treatment periods to confirm that BU08028 is safe for people. In addition, Dr. Ko and his team plan to investigate other compounds that stimulate both the MOR and the NOR, and might yield effects that are equally or more advantageous than those of BU08028. Dr. Ko says, “Ultimately, we would like to see one or two such MOR ligands to complete IND (Investigational New Drug)-enabling studies and eventually be established as a safe, nonaddictive analgesic to advance human medicine.”
Structure-Based Medication Discovery Bears Fruit
The laboratories of Drs. Bryan Roth from the University of North Carolina, Brian Shoichet from the University of California, San Francisco, Brian Kobilka from Stanford University, and Peter Gmeiner from Friedrich Alexander University took as their starting point for medication discovery detailed knowledge of the structure of the MOR that has recently been achieved through x-ray crystallography studies. These studies indicate that when morphine interacts with the MOR, it initiates two separate intracellular signaling pathways (see Figure 2). One pathway, originating with the G-coupled protein Gi, appears to underlie the medication’s valuable pain-relieving effect. The other, involving the protein β-arrestin, appears responsible for the medication’s dangerous respiratory and reinforcing effects.
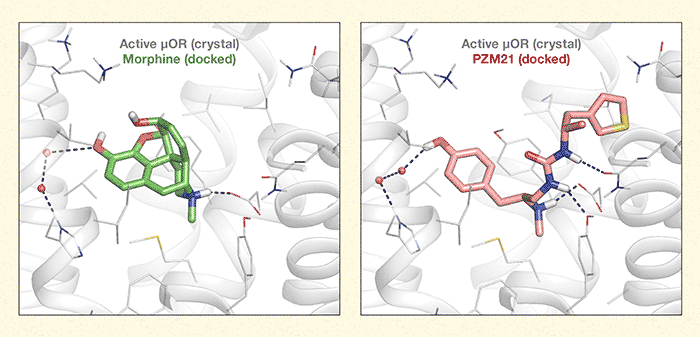 Figure 2. One Receptor, Two Ligands, Different Effects Morphine and PZM21 dock differently in the “binding pocket” of the mu opioid receptor (µOR), as indicated by the dotted lines. As a result of the docking differences, in mice, morphine promoted analgesia with undesirable effects of drug reinforcement, constipation, and respiratory depression; while PZM reduced the affective component of pain with little or none of morphine’s undesirable effects. Image courtesy of Dr. Anat Levit, Department of Pharmaceutical Chemistry, University of California, San Francisco.
Figure 2. One Receptor, Two Ligands, Different Effects Morphine and PZM21 dock differently in the “binding pocket” of the mu opioid receptor (µOR), as indicated by the dotted lines. As a result of the docking differences, in mice, morphine promoted analgesia with undesirable effects of drug reinforcement, constipation, and respiratory depression; while PZM reduced the affective component of pain with little or none of morphine’s undesirable effects. Image courtesy of Dr. Anat Levit, Department of Pharmaceutical Chemistry, University of California, San Francisco.
- Text Description of Figure 2
-
The figure illustrates how two different but related molecules—morphine and PZM21—dock to the same mu opioid receptor (μOR). The interaction of the receptor with morphine is shown in the left panel and the interaction with PZM21 in the right panel. Gray spirals and lines indicate structures of the active μOR as determined by computer modeling. The μOR structure is identical in both panels. The left panel shows in the center a green model of a morphine molecule docked at the μOR. The molecule consists of a core of several pentagons and hexagons with some arms sticking out from this core; some sections are highlighted in orange, blue, and white. Dotted lines leading from the morphine molecule to the left and bottom right indicate that some of these arms interact with specific atoms on the μOR. The right panel shows in the center a pink model of PZM21 docked in the active μOR. The molecule consists of a hexagon on the left end and a pentagon on the right end, which are connected by a chain-like structure with some arms sticking out to the left and right. Certain sections are highlighted in orange, blue, white, and yellow. Again, dotted lines from some of these arms to the left, bottom right, and right indicate interactions with specific atoms on the μOR. These interactions differ from those of morphine with the μOR, which may account for the different effects of morphine and PZM21.
The team set out to find a molecule that would engage the Gi pathway strongly and the β-arrestin pathway weakly if at all. Theoretically, such a molecule could equal or surpass morphine as a pain reliever and be very safe.
Using computational modeling, the researchers determined how the shapes of each of over 3 million commercially available compounds matched up with the docking site of the MOR. From among these, they identified 23 compounds that fit into the MOR, fit differently from morphine-based opioids, and, crucially, were chemically novel, unrelated to all known MOR ligands. Ultimately, the researchers chose one of these compounds and, after ascertaining that it activates the Gi pathway and not the β-arrestin pathway, and tinkering with its molecular structure to enhance its chances of success, they evaluated it in mice.
Mice treated with the compound, dubbed PZM21, remained on a hot plate about 87 percent longer than untreated mice before exhibiting signs of discomfort—either licking a paw or jumping off the plate. However, PZM21 did not increase the amount of time a mouse would tolerate a heat beam aimed at its tail before becoming uncomfortable enough to flick the tail out of the beam. The researchers interpret these mixed results as indicating that PZM21 reduces the emotional component of pain, but not the component that is simply reflexive. The analgesia provided by PZM21 lasted three times as long as that provided by morphine.
In addition, mice treated with PZM21:
- Did not exhibit behaviors that typically signify experiencing rewarding effects, such as increased locomotion or a special attraction to the chamber area where they had received the compound.
- Developed no respiratory depression and only mild constipation (another side effect of morphine and other traditional opioids).
Drs. Roth and Shoichet say that the main import of their work is that it demonstrates a way that knowledge of receptors’ crystal structures can be exploited to efficiently discover molecules with novel pharmacological properties and effects. Dr. Roth explains, “The overall approach provides a template for discovering novel drug-like molecules for an array of targets of opportunity for treating substance use disorders.” For example, they point out that no previously discovered opioid is as selective as PZM21 for the MOR’s Gi-coupled pathway, nor has any other opioid been reported to suppress the emotional component of pain and not the reflexive component. The investigators emphasize that the strategy can be used to develop medications that act through other receptors as well. (And, for example, NIDA-supported researchers recently mapped the crystal structure of the cannabinoid 1 receptor, the main site through which marijuana exerts its psychoactive effects.)
With respect to further development of PZM21, Dr. Roth says that, while PZM21 did not produce signs of abuse liability in locomotor stimulation or conditioned place preference tests, another important test remains: “Going forward, it will be important to determine if G-protein biased µ-opioid receptor agonists show less liability for self-administration.”
The study by Dr. Manglik and colleagues was supported by NIH grants GM106990 DA036246, GM59957, DA017204, and DA035764, and the work by Dr. Ding and colleagues was supported by NIH grants DA023281, DA032568, and DA035359.
Sources:
Ding, H.; Czoty, P.W.; Kiguchi, N. et al. A novel orvinol analog, BU08028, as a safe opioid analgesic without abuse liability in primates. Proceedings of the National Academy of Sciences of the United States of America 113(37):E5511-E5518, 2016. Abstract
Manglik, A.; Lin, H.; Aryal, D.K. et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 537(7619):185-190, 2016. Abstract