
- Investigators have shown that 2-AG, an endocannabinoid (i.e., a cannabinoid manufactured within the body, as opposed to plant-derived), augments the cocaine-induced dopamine surge in the brain’s reward system.
- The discovery adds to evidence that inhibiting activity in the endocannabinoid system might reduce cocaine’s rewarding and addictive effects.
Cocaine, like most drugs that produce rewarding and addictive effects, does so by abruptly and massively increasing levels of the neurotransmitter dopamine in the brain’s nucleus accumbens (NAc). Cocaine makes dopamine surge in the NAc primarily by blocking the dopamine transporter, a molecule that removes excess dopamine from circulation. However, recent NIDA research reveals that a second mechanism accounts for a significant portion of cocaine-induced dopamine surfeit in the NAc.
The principal player in the newly discovered mechanism is the endocannabinoid molecule 2-arachidonoylglycerol (2-AG). In the new research, Drs. Huikun Wang and Carl Lupica of NIDA’s Intramural Research Program, with colleagues from NIDA and the University of Maryland, found that:
- Cocaine induced dopaminergic (dopamine-releasing) neurons to step up production of 2-AG.
- Treating rats with a compound (rimonabant) that inhibits 2-AG activity before injecting the animals with cocaine cut cocaine-induced increases in NAc dopamine concentrations by approximately 50 percent.
Inhibitor Inhibited
Dopaminergic neurons release dopamine from axon terminals located in the NAc. How much dopamine they release depends on their firing intensity, which in turn depends on:
 Figure 1. Dopaminergic Neurons in the Brain Dopaminergic neurons reside in the ventral tegmental area (VTA). When stimulated, they release dopamine from their axon terminals in the nucleus accumbens (NAc).
Figure 1. Dopaminergic Neurons in the Brain Dopaminergic neurons reside in the ventral tegmental area (VTA). When stimulated, they release dopamine from their axon terminals in the nucleus accumbens (NAc).
- Text Description of Figure 1
-
The figure shows a schematic of the location of dopaminergic neurons in the brain. The brain is shown as a length-wise cross-section, with various brain regions outlined. A red neuron indicates the location of the dopaminergic neurons. The cell body of this neuron is located in the middle of the lower part of the brain in an area labeled VTA (ventral tegmental area). A long extension from the neuron’s cell body—the neuron’s axon—extends to the left (i.e., towards the front of the brain), to an oval region outlined in green and labeled NAc (nucleus accumbens). There, the neuron splits into several ends, the axon terminals where the dopamine is released.
- How much stimulatory input they receive from glutamatergic neurons in another brain region, the ventral tegmental area (VTA) (see Figure 1).
- How much inhibitory input they receive from gamma-aminobutyric (GABA) neurons in the VTA.
Drs. Wang and Lupica demonstrated that cocaine mobilizes the endocannabinoid system to reduce GABA inputs to dopaminergic neurons. In experiments with rats, they showed that cocaine precipitates the release of 2-AG into the VTA synapses between GABAergic neurons and dopaminergic neurons. The 2-AG interacts with cannabinoid receptors on the GABAergic neurons, slowing the neurons' firing and reducing their release of the neurotransmitter.
The researchers elucidated the complete molecular pathway whereby cocaine reduces GABA release in the VTA and increases dopamine release in the NAc (Figure 2):
- Cocaine blocks noradrenergic neurons from clearing the neurotransmitter norepinephrine from circulation in the VTA.
- Norepinephrine levels consequently rise, stimulating α1 receptors on VTA dopaminergic neurons.
- Activation of the α1 receptors triggers reactions within the dopaminergic neurons that culminate in the release of 2-AG.
- 2-AG binds to cannabinoid (CB1) receptors on GABAergic neurons.
- 2-AG’s interaction with the CB1 receptor reduces GABAergic neurons’ release of GABA.
- Less exposure to GABA, an inhibitory neurotransmitter, frees the dopaminergic neurons to fire more intensely and release more dopamine into the NAc.
Figure 2. The Role of 2-AG in Enhancing Cocaine Reward
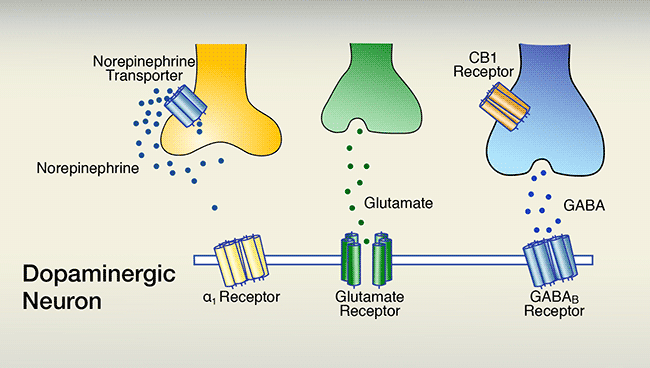
A. The firing of dopaminergic neurons that are situated in the ventral tegmental area underlies feelings of reward. Dopaminergic-neuron firing is promoted by synaptic glutamate activating glutamate receptors, and inhibited by synaptic gamma-aminobutyric acid (GABA) activating GABAB receptors. A third neurotransmitter, norepinephrine, which can indirectly promote dopaminergic firing by activating α1 receptors, generally is inactive because it is taken back up into its releasing cell via the norepinephrine transporter.
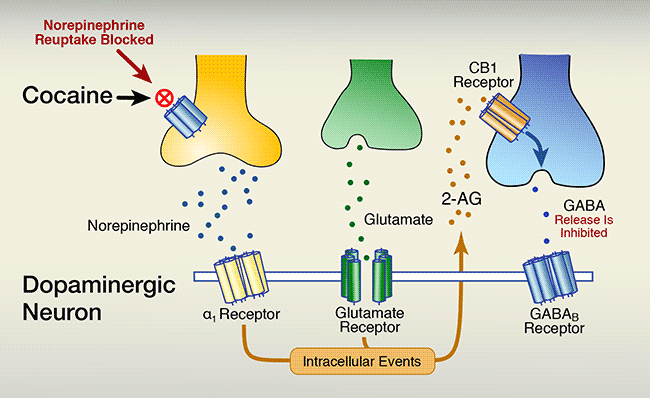
B. Cocaine blocks the norepinephrine transporter from drawing the neurotransmitter back into its releasing cell. Consequently, norepinephrine builds up in the synapse. Higher concentrations of synaptic norepinephrine stimulate G-protein–coupled α1 receptors to:
- increase biochemical processes that release 2-AG, and
- increase dopamine neuron activity, further increasing 2- AG, which;
- enhances activation of cannabinoid-1 (CB1) receptors on GABA neurons, which;
- lowers GABA neurons’ release of GABA into the synapse, which;
- tilts the balance of glutamate stimulation and GABA inhibition of dopaminergic neuron firing in favor of stimulation, which;
- increases the neurons’ release of dopamine into the nucleus accumbens, which;
- enhances feelings of reward.
- Text Description of Figure 2
-
The figure consists of 2 panels, both of which show a schematic representation of a section of a dopamine-releasing (i.e., dopaminergic) neuron and of three other neurons that interact with the dopaminergic neuron. The membrane of the dopaminergic neuron is indicated by a horizontal double line. Integrated into the membrane are three different receptors, indicated as structures consisting of several rod-like subunits: a yellow α1 receptor, a green glutamate receptor, and a blue gamma-aminobutyric acid (GABAB) receptor. The α1 and GABAB receptors have a similar shape, indicating that they act through a similar mechanism of action; the glutamate receptor has a different shape, indicating it has a different mechanism of action. Above the dopaminergic neuron are a yellow neuron that releases the neurotransmitter norepinephrine, indicated by dark blue dots; a green neuron that releases the neurotransmitter glutamate, indicated by green dots; and a blue neuron that releases GABA, indicated by light-blue dots. The blue GABA neuron also carries a receptor called cannabinoid 1 (CB1) receptor, indicated in brown. The yellow neuron also carries a norepinephrine transporter (indicated in blue) that can move norepinephrine from the space between the cells back into the norepinephrine-releasing neuron.
The upper panel shows the situation in the absence of cocaine. Norepinephrine released by the yellow norepinephrine neuron stays near that neuron and is transported back into the cell via the norepinephrine transporter; very little, if any, norepinephrine reaches the α1 receptor. Glutamate released from the green glutamate neuron and GABA released from the blue GABA neuron both reach their corresponding receptors on the dopaminergic neuron. Glutamate is a stimulatory neurotransmitter that promotes dopamine release by the dopaminergic neuron, whereas GABA is an inhibitory neurotransmitter that reduces dopamine release. The combined actions of glutamate and GABA result in a certain level of dopamine release.
The lower panel shows the situation in the presence of cocaine. Cocaine, indicated by a red circle with a cross in it, binds to the norepinephrine transporter on the yellow norepinephrine neuron, blocking the transport of norepinephrine back into the cell. Instead, the norepinephrine is distributed in the space between the cells and interacts with the α1 receptor on the dopaminergic neuron. At the same time, glutamate released from the green glutamate neuron still reaches the glutamate receptor on the dopaminergic neuron. The interactions of norepinephrine and glutamate with their corresponding receptors together trigger intracellular events in the dopaminergic neuron—indicated as a brown box—that result in the production and release of the endocannabinoid 2-arachidonoylglycerol (2-AG)—indicated as brown dots. The 2-AG molecules interact with the brown CB1 receptor on the blue GABA neuron. A blue arrow within the neuron indicates that this interaction inhibits the release of GABA from the neuron. Fewer GABA molecules than without cocaine are found in the space between the cells and can interact with the GABAB receptor. As a result of the cocaine-induced actions of norepinephrine and the continued activation by glutamate, coupled with the reduced activity of GABA resulting from the cocaine-induced intracellular events, the dopaminergic neuron can release more dopamine.
A Treatment Approach?
2-AG is one of several neurotransmitters in the endocannabinoid system, which serves a wide variety of regulatory functions in the brain and body. The system has previously been implicated in drugs’ rewarding effects: for example, the marijuana ingredient tetrahydrocannabinol (THC) produces its “high” by infiltrating and activating the endocannabinoid system. Some studies with animals have tried using medications that block cannabinoid receptors to alleviate drug-seeking behaviors, with encouraging results.
Drs. Wang and Lupica’s findings further advance this body of research, and provide a more detailed picture of the role of endocannabinoids in cocaine use. The researchers note, however, that the 2-AG pathway does not fully account for cocaine-induced increases in dopaminergic firing intensity: The neurotransmitter serotonin also contributes. When the researchers administered cocaine together with compounds that increase serotonin levels in the VTA, they observed an inhibitory effect on GABA neurons similar to that produced by 2-AG. Moreover, blocking cannabinoid activity did not completely abolish cocaine’s effect on GABA unless serotonin activity was also blocked (see Figure 3).
In summary, Dr. Lupica says, “It appears that the endocannabinoids mediate some of cocaine’s effects within the brain’s reward circuitry. If the rewarding and addictive properties of cocaine require intact endocannabinoid function, then disrupting this function with anti-cannabinoid drugs may help to reduce these properties of cocaine. Through these types of studies, we may be able to develop a useful tool to combat cocaine addiction.”
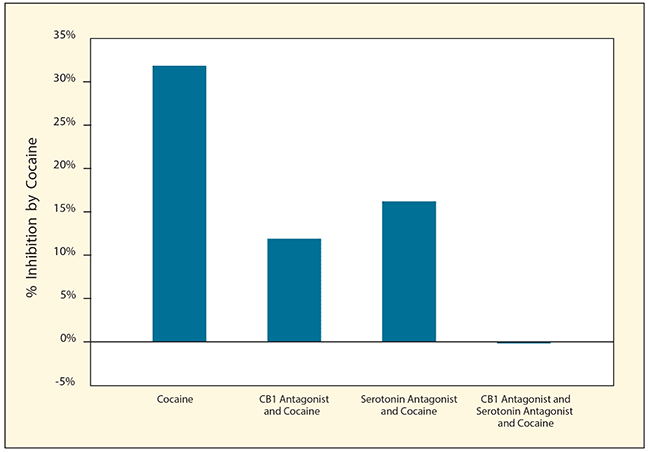 Figure 3. Blocking Endocannabinoids and Serotonin Prevents Cocaine From Inhibiting GABA Cocaine reduced the inhibitory effects of GABA on dopamine neurons in living rats. Cocaine’s effects on GABA were reduced when animals were treated with either a CB1 antagonist or a serotonin antagonist, and completely blocked by treatment with both antagonists.
Figure 3. Blocking Endocannabinoids and Serotonin Prevents Cocaine From Inhibiting GABA Cocaine reduced the inhibitory effects of GABA on dopamine neurons in living rats. Cocaine’s effects on GABA were reduced when animals were treated with either a CB1 antagonist or a serotonin antagonist, and completely blocked by treatment with both antagonists.
- Text Description of Figure 3
-
The figure shows a bar graph demonstrating the effects of cocaine with and without CB1 antagonists, serotonin antagonists, or both on the inhibitory effects of GABA on dopaminergic neurons in living rats. The horizontal (y-) axis shows the percent inhibition of GABA activity by cocaine. The left bar shows that treatment of rats with cocaine only inhibits GABA activity by approximately 32 percent. The second bar shows that treatment with cocaine plus a CB1 antagonist reduces the inhibition to about 12 percent, and the third bar shows that treatment with cocaine plus a serotonin antagonist reduces the inhibition to about 17 percent. The right bar shows that treatment with cocaine plus both a CB1 antagonist and a serotonin antagonist completely eliminates inhibition of GABA activity by cocaine.
This study was supported by NIH grants DA022340 and DA033926, and by the NIH/NIDA Intramural Research Program.
Source:
Wang, H.; Treadway, T.; Covey, D.P.; Cheer, J.F.; Lupica, C.R. Cocaine-Induced Endocannabinoid Mobilization in the Ventral Tegmental Area. Cell Reports 12(12):1997-2008, 2015. Full article