

In 2017, Dr. Linda Dwoskin applied to NIDA's Grand Opportunity for Medications Development program for support to continue her quest to develop the first-ever medication to reduce craving and relapse in methamphetamine use disorder. The goal of her project aligned perfectly with the program's objective "to fund medication studies that will have a high impact and quickly yield the necessary results to advance medication closer to FDA approval." Success would fill an enormous need, affecting nearly 700,000 Americans with problematic use of the stimulant. Moreover, Dr. Dwoskin and her team at the Universities of Kentucky and Arkansas had brought their work to a place where they could hope that one last push might put them over the finish line.
While Dr. Dwoskin waited to learn the fate of her grant application, she worried. Giving patients and treatment providers a weapon against the ravages of methamphetamine addiction would mean more to her, professionally and personally, than anything else she had accomplished in her distinguished career as researcher and educator. But without new funding, she would have scant hope of ever achieving this goal to which she had devoted 20-plus years of diligent, concentrated effort. (Read Parts 1, 2, and 3 of this article series.)
If NIDA turned down Dr. Dwoskin's application, she might be able to get money elsewhere, but that would be a long shot. She says, "We've had some sniffing around from private companies that might be interested in providing some funding, because we're pretty close to our goal of a molecule that meets the criteria for testing in people. But they would have to see positive data before investing, and I would have to secure funding somewhere to produce the positive data."
Meanwhile, Dr. Dwoskin appealed to her institution, the University of Kentucky, which provided her with interim funding. "The university was confident based on our track record that we would succeed eventually," says Dr. Dwoskin. These funds, plus some money she had remaining from her previous NIDA grant, enabled her to keep the work moving, albeit at a reduced level of activity.
"The most important thing was to keep the team together," she says. "We have six technical staff who are the workhorses on the project. They perform the assays, analyze the data, present their findings in team meetings, and contribute ideas for trouble shooting problems that arise. Maintaining their involvement and efforts is critical, especially for our project where day-to-day consistency in performance of the assays is needed." With the interim funds in hand, Dr. Dwoskin could pay her technical staff salaries and continue to support graduate students, some of whom were relying on project data for their Ph.D. theses. Faculty members donated time during this lean period.
A String Attached
NIDA awarded Dr. Dwoskin a 2-year grant. As a condition for receiving the second year's disbursement, she and her team would have to crack the problem of bioavailability, which is a measure of how much of an ingested drug makes it to the blood circulation.
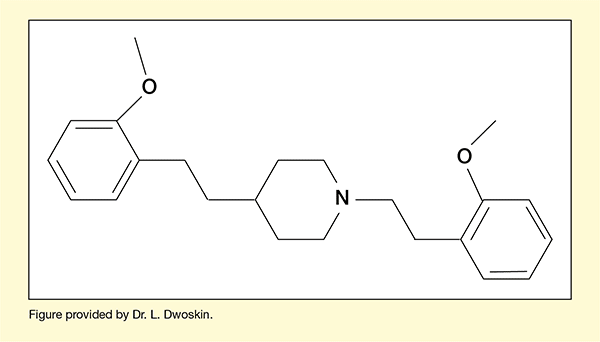
The team had developed a molecule, JPC-077, that performed very well in comprehensive in vitro testing and demonstrated efficacy in reducing methamphetamine-related behaviors in animal testing (see Figure 1). The team were optimistic about their chances of advancing JPC-077 to the developmental stage of clinical testing in people, but first they needed to fix a shortcoming. The molecule had oral bioavailability of only about 3 percent, meaning that less than 3 percent of an oral dose made it into the systemic blood circulation. This was unacceptable because only the portion of the drug that entered the systemic circulation would be carried to the site in the brain where the molecule exerts its therapeutic activity.
The terms of the NIDA grant set for the team a milestone of reaching 30 percent bioavailability by the end of the first funded year (see Bioavailability Measurement). Dr. Mark Leggas, the team's lead pharmacokineticist, explains, "A rule of thumb is that a drug needs at least 30 percent bioavailability to reliably confer therapeutic effects. If you are below 30 percent, there is variability in how people absorb a drug. For example, if a drug has an average bioavailability of 5 percent, some people might absorb 10 percent of a dose into their bloodstream and others only 1 percent." The drug might be effective in the high absorbers, but not the low absorbers. "This is a much bigger difference than the case where a drug has 30 percent bioavailability with a range of 25–35 percent."
Vicissitudes of Drugs
The team set out to learn what was keeping more than 90 percent of a dose of JPC-077 out of animals’ systemic circulation. Once they knew this, they would try to modify the molecule to avert the problem.
"When you take a drug orally, many things can happen to it," says Dr. Leggas. It may be broken down in the stomach or in the intestine, or it may just precipitate and never be absorbed. Both the gastrointestinal acidity and the intestinal flora can play a role in drug stability. Drugs that traverse the stomach and intestine intact funnel into the portal vein that flows into the liver, which acts like a filter, either removing or breaking down toxins and drugs that get absorbed. Drug molecules that escape the liver enter the systemic circulation and may reach their site of action.
Dr. Leggas and his colleagues worked through these possibilities and found that 6 hours after ingesting a dose, mice had neither the intact JPC-077 nor metabolites in their stomach or intestine. These findings indicated that the molecule survived the gastrointestinal tract and was metabolized in the liver. “That's actually a common scenario," says Dr. Leggas. "Most drugs are broken down in the liver. The liver contains the majority of enzymes for processing nutrients and clarifying toxins before they reach the systemic circulation."
A Culprit Enzyme
The next question for the researchers: Which of the wide panoply of liver enzymes were metabolizing JPC-077? Based on the structural features of JPC-077, the Kentucky team thought that the liver enzyme CYP2D6 (see Figure 2) was likely responsible for the poor bioavailability of JPC-077. This enzyme is known to contribute to the metabolism of many drugs.
Dr. Leggas fed information about JPC-077 and CYP2D6 into a computer model, which predicted that the two did indeed interact. To confirm this result in vivo, the researchers gave JPC-077 to rats and used mass spectroscopy to analyze the animals' blood for the metabolites that result when CYP2D6 breaks down JPC-077. Metabolites were present. Moreover, they were present in large enough quantities to account for a large portion of the JPC-077 that never made it into the systemic circulation.
From 10 to 40
The finding that CYP2D6 was the main metabolizer of JPC-077 highlighted the importance of enhancing the molecule's bioavailability. Dr. Leggas explains, "The CYP2D6 enzyme comes in four different forms. Depending on which form people have, they will be ultra-rapid metabolizers of a drug, rapid metabolizers, intermediate metabolizers, or slow metabolizers." Establishing dosage levels that both ultra-rapid and slow metabolizers respond to can be difficult, all the more so when overall bioavailability is low. In such a case, says Dr. Leggas, "You would likely have to perform genetic testing beforehand to make sure that you give a patient the right dose."
Dr. Dwoskin's team set out to modify the structure of JPC-077 to prevent it from being metabolized by CYP2D6 before entering the systemic circulation. Medicinal chemist Dr. Peter Crooks at the University of Arkansas for Medical Sciences generated a series of JPC-077 molecules with alterations to the chemical components that interact with the enzyme. "Understanding how the molecule was metabolized allowed us to make rational decisions for improving its structure," says Dr. Leggas.
First, Dr. Crooks and his laboratory colleagues tried modifying JPC-077 in a way that scientists working at pharmaceutical companies had used successfully to reduce CYP2D6 metabolism and increase bioavailability of some of their companies' products. The modification slowed metabolism of JPC-077, but it did not raise bioavailability to the required 30 percent.
The team's next modification strategy for JPC-077 produced a variant that met the bioavailability requirement. Dr. Leggas says, "We now have a molecule that looks like JPC-077 and gives us bioavailability roughly in the 40 percent range." Unfortunately, the modifications that yielded this leap in bioavailability also weakened the molecule's counteracting effect on methamphetamine at its target site (the ventricular monoamine transporter 2).
By the terms of their NIDA grant, Dr. Dwoskin and her team have until July to produce a molecule with both the efficacy at the target and other in vitro and in vivo standards that JPC-077 had met, and the 30 percent bioavailability they achieved with the recent variant.
Dr. Dwoskin says, "We definitely are approaching this with guarded optimism." She shares an anecdote: "When Mark presented the data that we had reached 40 percent bioavailability to our scientists and technicians and graduate students, everybody just sat there. One faculty member asked, 'How come everybody's not more excited?' And someone chimed up, ‘Well, we've been burned before. We're waiting for the next shoe to drop.' "
She continues, "It feels like we're closing in on our goal. The team has amassed a lot of knowledge and they are leveraging it by building computational models. That and the experience of our medicinal experts and pharmacologists should help us find the right path to overcome the remaining hurdles. Theoretically, we should be able to take a molecule into the clinic for testing in people in 2 years. But in this business unanticipated problems tend to arise, so realistically I think it may take three."
Bioavailability Measurement
A medication's bioavailability is essentially the portion of a dose that enters the systemic circulation. The bioavailability of an intravenous dose of a medication is considered to be 100 percent, since the medication is administered directly into the bloodstream. An oral medication generally has lower bioavailability, since some is absorbed, metabolized, or otherwise eliminated before it reaches the bloodstream. A medication’s oral bioavailability is often reported as a percentage of its intravenous bioavailability.
To estimate the oral bioavailability of JPC-077 variants, Dr. Leggas and colleagues measured concentrations of the molecule in rats' blood at serial time points following intravenous and oral doses. The measurements, made with mass spectroscopy, were used to conduct pharmacokinetic analyses which revealed that one variant's oral bioavailability was less than 10 percent of its intravenous bioavailability, and another's was over 40 percent. Ten percent bioavailability was too low to qualify the first variant for further consideration as a potential medication. The second variant’s bioavailability was acceptable, but the molecule had a different shortcoming that forced the scientists to keep searching.